
Explications générales
Une video pour les enfants
Quel est la cause du syndrome de Marfan ?
Le syndrome de Marfan est lié à une mutation génétique, retrouvée le plus souvent dans le gène FBN1. Nous possédons tous deux copies de ce gène, une copie reçue de chacun de nos parents.
Une mutation dans le gène FBN1 entraine un manque de production de la Fibrilline rendant les tissus conjonctifs trop élastiques. Cette hyper-élasticité est la cause des signes retrouvés dans le syndrome de Marfan.
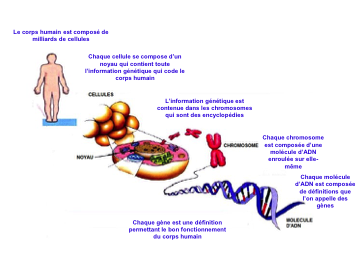
Qu’est-ce qu’un gène ?
L’être humain est définit par environ 30000 gènes.
Ces gènes sont des définitions qui permettent le bon fonctionnement et la structure du corps humain.
Chaque gène est en double exemplaire, un exemplaire hérité de notre père et l’autre de notre mère.
Qu’est-ce qu’une mutation génétique ?
Une mutation génétique est comme une faute d’orthographe dans l’écriture du gène entrainant une mauvaise compréhension par la cellule.
Un film qui explique la génétique et le séquençage, fait par la Filière de Santé AnDDI-Rares.
Quel est le risque de transmettre le Syndrome de Marfan ?
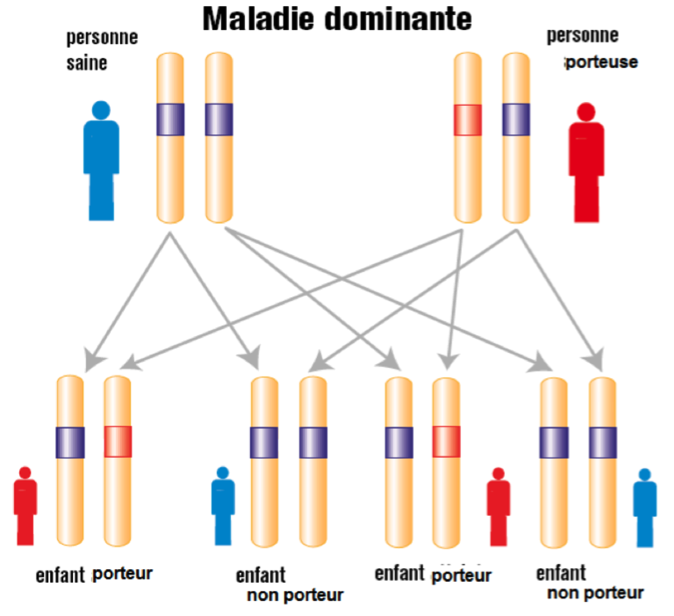
Une mutation génétique entrainant un syndrome de Marfan n’est portée que par une seule des deux copies du gène en cause. On parle de transmission autosomique dominante.
Nous transmettons à chacun de nos enfants la moitié de notre patrimoine génétique. Une personne atteinte du syndrome de Marfan a donc un risque sur deux de le transmettre à sa descendance et une chance sur deux de ne pas le transmettre et ce pour chaque enfant.
Que signifie autosomique dominant ?
La transmission est de type autosomique dominant :
- autosomique signifie que la pathologie touche autant les hommes que les femmes puisque la mutation est portée par les chromosomes non sexuels appelés autosomes.
- Dominant, signifie qu’il y a un risque sur deux de transmettre la mutation a chacun de ses enfants (fille ou garçon) car la mutation est portée par une seule des deux copies du gène.
Pourquoi y-a-t-il une grande diversité de signes cliniques dans le syndrome de Marfan ?
Le gène FBN1 qui est le plus fréquemment impliqué dans le syndrome de Marfan, permet de fabriquer une protéine, la Fibrilline, qui est présente dans pratiquement tous les tissus du corps. Ceci explique la grande diversité de signes cliniques rencontrés (cœur, œil, squelette).
Quel rapport entre le gène et les signes retrouvés au niveau du corps ?
Le syndrome de Marfan est causé par un manque d’une protéine, appelée Fibrilline, dans les tissus conjonctifs qui se retrouvent partout (peau, cœur, œil, tendons, etc.). Cette protéine est en manque dans les tissus car au niveau du gène qui code pour cette protéine (gène que nous possédons tous) il y a une faute d’orthographe, appelée mutation, présente sur une seule des deux copies de ce gène.
La copie du gène qui ne porte pas la faute d’orthographe produit de la Fibrilline et la copie qui porte la faute d’orthographe n’en produit pas ce qui entraine un manque de Fibrilline au niveau des tissus conjonctifs, ce qui les rend trop élastiques et entraine les signes cliniques du syndrome de Marfan.
Peut-il y avoir des atteintes différentes dans une même famille et pourquoi ?
Il y a des atteintes cliniques différentes au sein d’une même famille malgré le fait que ce soit la même mutation chez tout le monde.
Ceci est probablement dû aux restes de nos gènes qui sont forcément différents de notre frère, de notre mère ou encore de notre père, puisqu’on hérite de la moitié des gènes de son père et de la moitié des gènes de sa mère.
Les facteurs environnementaux tel que nos activités, notre métier, notre façon de vivre pourraient expliqués une partie de ces différences.
Peut-on prévoir les atteintes cliniques ?
Nous ne pouvons pas actuellement prédire la nature exacte des signes cliniques d’un individu à l’autre porteur de la même mutation génétique.
Les hommes et les femmes sont ils atteints de la même façon?
La probabilité d’atteinte en terme de transmission génétique est la même chez les hommes et les femmes.
Comment fait-on pour trouver le gène responsable du syndrome de Marfan
Comment trouve-t-on une mutation génétique ?
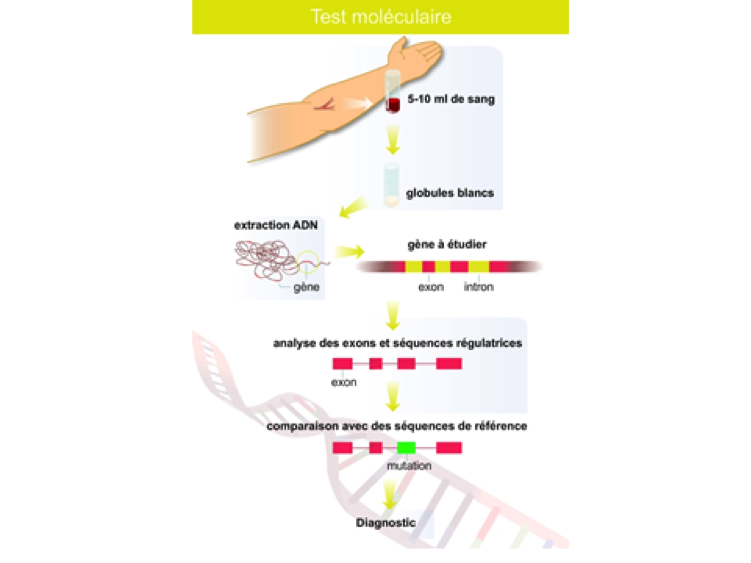
Pour entamer une analyse génétique, il suffit d’une prise de sang, à partir de laquelle nous isolons l’ADN (ADN= stockage de l’information génétique et qui est identique dans toutes les cellules de l’organisme). L’ADN est ensuite extrait afin de pouvoir lire les gènes impliqués dans le syndrome de Marfan.
Les résultats obtenus seront comparés aux séquences de références. Si des différences sont observées le biologiste évalue les conséquences qu’elles auront sur le fonctionnement du gène et de la protéine fabriquée. Il en tire une conclusion qui est transmise au prescripteur, prescripteur qui rend en mains propres le résultat, en expliquant au patient ce que cela implique pour lui et sa famille.
Combien de temps faut-il pour obtenir les résultats d’un test génétique dans le cadre du syndrome de Marfan ?
Il faut environ deux à trois ans pour obtenir un résultat d’analyse génétique. Ce temps s’explique par le fait qu’il n’y ait qu’un seul laboratoire qui effectue ces analyses en France, étant donné que nous sommes dans le cadre d’une maladie rare.
Les évolutions technologiques actuelles laissent entrevoir une rapide amélioration des délais à court terme.
Quand peut-on faire le test génétique?
On peut faire le test génétique principalement dans 2 situations :
- lorsque le diagnostic de syndrome de Marfan est porté sur des signes cliniques p4 ou fortement suspecté (diagnostic en attente) et dans ce cas le but est d’identifier une anomalie génétique en cause. L’intérêt du test est double : confirmer le diagnostic et faciliter le dépistage familial.
- lorsqu’une mutation familiale est déjà connue, les tests visant à rechercher cette mutation sont proposés aux apparentés.
Si le test est réalisé sans examen clinique, un second prélèvement est obligatoire.
Pourquoi faut-il faire 2 prélèvements pour l’étude génétique ?
Nous vérifions sur deux prélèvements distincts le résultat génétique lorsque nous n’avons pas examiné les patients dans le cadre d’une enquête familiale ou lorsque le résultat diverge de l’examen clinique.
Ceci dans le but de s’assurer qu’il n’y ait pas eu d’erreur d’analyse.
Que faire du résultat de génétique ?
Le résultat de génétique moléculaire est strictement confidentiel. Il ne doit être connu que des médecins prenant en charge directement votre syndrome de Marfan (centre de référence/compétence ; médecin généticien).
Nous vous recommandons d’informer vos médecins que vous avez un syndrome de Marfan, afin de vous assurer la meilleure prise en charge possible.
A quoi sert le résultat génétique ?
Le résultat génétique permet de trouver le gène impliqué dans les signes cliniques du Syndrome de Marfan et d’orienter ainsi la prise en charge.
Il facilite également l’enquête familiale.
Que faire du résultat de génétique ? Faut-il le mentionner dans le carnet de santé ? En parler à son employeur, son assureur ?
Les analyses génétiques sont propres à la personne et n’ont aucune valeur juridique.
Personne n’est en droit de vous demander ces résultats.
Nous vous conseillons donc de ne pas le mentionner dans le carnet de santé ou à votre employeur/assureur car cela pourrait vous porter préjudice.
La loi de bioéthique de 2004 et la convention AERAS protègent vos droits. Vous n’avez à déclarer que la prise en charge de vos signes cliniques.
http://www.aeras-infos.fr/webdav/site/aeras/shared/contents/3-Publications/Rapports_des_instances/Annexe_2_-_Loi_2007-131_du_31_janvier_2007.pdf
Est-ce que le syndrome de Marfan est toujours hérité d’un des deux parents ?
Dans environ 1/3 des cas une personne atteinte du syndrome de Marfan a deux parents indemnes. Dans ce cas, une néo-mutation (nouvelle mutation) s’est produite spontanément soit dans un ovule de la mère, soit dans un spermatozoïde du père soit précocement après la fécondation pendant le développement embryonnaire. Pour la personne atteinte le risque de transmettre la maladie a la descendance est le même que dans les formes familiales.
Est ce que la mutation peut sauter une génération ?
Non, d’un point de vue génétique (càd la transmission de la mutation du gène FBN1) cela n’existe pas. Le gène ne peut pas sauter une génération.
Par contre la variabilité d’expression de la maladie au sein même d’une famille peut donner l’impression de saut de génération sur une base clinique quand un sujet porteur de la mutation est atteint de façon suffisamment discrète pour que cela passe inaperçu à des yeux non experts. Une personne ayant la mutation génétique, peut ne présenter aucun symptôme, alors que son enfant en présentera plus (ou inversement).
C’est une des raisons pour laquelle, lorsque la mutation familiale est connue, il est toujours préférable de la rechercher dans toute la famille, même auprès de ceux qui n’ont aucun signe clinique.
Que peut-on faire pour ne pas transmettre une mutation génétique à sa descendance ?
Cette question est très personnelle et la réponse est propre à chacun.
Le risque de transmettre une mutation génétique entrainant un syndrome de marfan est de un sur deux pour chaque enfant.
Il est possible d’avoir recours à l’adoption ou au don de sperme (si c’est l’homme qui est atteint).
Si, et seulement si, une mutation génétique est mise en évidence, l’autre alternative est la procréation médicalement assistée (PMA). Par PMA on entend :
- le diagnostic prénatal (DPN)
- le diagnostique pré-implantatoire (DPI = bébé éprouvette)
L’assistance médicale à la procréation est discutée au cas par cas auprès d’un conseil pluridisciplinaire de diagnostic prénatal (CPDPN).
Le risque de transmettre le syndrome aux enfants est-il le même pour un homme ou une femme atteint?
Oui, le risque est le même car les gènes actuellement connus en cause dans le syndrome de Marfan sont situés sur les chromosomes appelés « autosomes » (chromosomes non sexuels) qui sont donc les mêmes chez l’homme et la femme.
Qu’est ce que l’assistance médicale à la procréation? Est-ce possible dans le cadre du syndrome de Marfan?
La procréation médicalement assistée (PMA), également appelée assistance médicale à la procréation (AMP), est un ensemble de pratiques cliniques et biologiques où la médecine intervient plus ou moins directement dans la procréation. Dans le cas du syndrome de Marfan, la PMA peut prendre différentes formes : insémination artificielle dans le cas d’un don de sperme, diagnostic anténatal durant une grossesse, diagnostic préimplantatoire avant la grossesse sur un embryon obtenu par fécondation in vitro.
Qu’est-ce que le diagnostic prénatal (DPN) ? Comment se déroulent-t-ils? Où peut-on les réaliser?
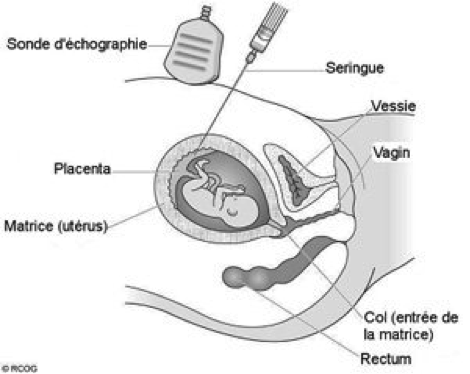
Le diagnostic prénatal peut être réalisé, si tel est le choix du couple, à partir d’une biopsie de trophoblaste pour déterminer si le fœtus est porteur ou non de la mutation. Cet examen est un geste invasif qui se pratique à partir de 11 à 12 semaines d’aménorrhées et qui implique un risque de fausse couche spontanée de 0.5 à 1%. Ce risque explique pourquoi un DPN n’est pas entrepris uniquement dans le but de connaître le statut du fœtus, mais bien s’il y a une volonté d’interruption médicale de grossesse si le fœtus s’avérait porteur de la mutation familiale. Une fois le prélèvement effectué une mise en culture de ces cellules de 15 jours est nécessaire avant de connaitre le résultat final. En fonction du résultat le couple décide ou non d’interrompre la grossesse si l’enfant était porteur.
Qu’est-ce que le diagnostic préimplantatoire (DPI)? Comment se déroulent-t-ils?
- Le diagnostic préimplantatoire consiste, après fécondation in vitro, à déterminer si les embryons sont porteurs ou non de la mutation. Le diagnostic préimplantatoire nécessite une fécondation in vitro et donc un prélèvement d’ovocytes chez la mère après stimulation ovarienne. Cette technique nécessite que la femme ait une réserve ovarienne importante et un âge inférieur à 38 ans au moment des examens.
- Si la réserve ovarienne est suffisante, une stimulation ovarienne par piqure d’hormones sera réalisée dans le but d’obtenir un maximum d’ovocytes.
- Les ovocytes sont ensuite prélevés par cœlioscopie.
- La cœlioscopie consiste à accéder à la cavité abdominale sans ouvrir la paroi abdominale.
- Ces ovocytes sont fécondés par les spermatozoïdes dans une éprouvette.
- On recueille au maximum 8 ovocytes fécondés.
- une fois les embryons développés au stade de 6-8 cellules, une cellule de chaque embryon est prélevée et analysée.
- La recherche de la mutation génétique est alors réalisée sur ces cellules afin de savoir si les embryons sont porteurs de la mutation.
- Pour continuer la technique, il faut qu’il y ait des embryons non porteurs de la mutation.
- On réimplante ensuite dans l’utérus un seul embryon non porteur. Les autres embryons non porteurs seront conservés.
Le taux de réussite de l’implantation est estimé entre 20 et 30%.
Par ailleurs, dans l’état actuel des moyens des centres réalisant le diagnostic pré implantatoire, les délais d’attente sont relativement long et il se passe souvent deux à cinq ans entre le premier rendez-vous au centre de diagnostic préimplantatoire et la mise en route d’une grossesse.
Il est important de mentionner qu’il existe un risque d’échec à chaque étape.
La fiabilité de cette technique n’est donc par conséquent pas de 100%
Le diagnostic prénatal (DPN) et le diagnostic préimplantatoire (DPI) sont-ils pris en charge par la sécurité sociale ?
Oui même si le nombre d’essai est limité pour le DPI.
Est ce que les techniques de diagnostic préimplantatoire (DPI) ou de diagnostic prénatal (DPN) sont fiables ?
La technique du DPN est fiable à 100%. En ce qui concerne le DPI il y a tout un risque d’échec à chaque étape de la technique, de plus comme l’analyse génétique est réalisée à partir d’une seule cellule, il y a donc une très faible quantité de patrimoine génétique ce qui rend difficile l’analyse et diminue la fiabilité.
A partir de quel age peut-on faire le test génétique? Est-on obligé de venir à Bichat?
Le test génétique peut être réalisé à partir de l’âge de 4 ans chez tous les apparentés au premier degré d’un sujet atteint.
Ce test peut-être réalisé dans tous centres de génétique en France après consultation auprès d’un généticien dans le cas d’un diagnostic familial.